20221104康测科技助力客户发表CMGH:原来高脂饮食引起脂肪肝变性的关键因素竟是它?
#
研究背景
非酒精性脂肪肝(NAFLD)是病人肝脏相关疾病发病率和死亡率较高的主要原因。脂肪滴的积累是由于肝脏脂质稳态不平衡造成的:通过脂肪酸摄取和脂肪新生获得的脂滴超过了通过脂肪酸氧化和脂质输出的处理(小编简单理解为脂肪摄入速度超过了脂肪消耗速度)。有研究表明,NAFLD的发病机制可能是由基因和环境因素(如营养过剩)之间的动态相互作用形成的,表观遗传学是一个机制桥梁。
DNA 6mA修饰近期被鉴定为表观遗传的marker,6mA修饰分布模式具有物种和组织特异性。其中ALKBH1为DNA 6mA修饰的去甲基化酶,那么营养过剩是如何触发表观遗传机制来影响NAFLD的进展呢?跟着小编一起看看下面这篇文章吧!
#
研究结果
1、肥胖小鼠肝脏中DNA 6mA水平升高
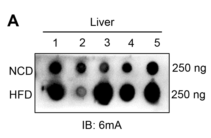
为了探讨DNA 6mA修饰与脂肪肝的关系,作者对肝脂肪变性的高脂饮食(HFD)小鼠肝脏中的DNA 6mA水平进行检测,结果显示HFD小鼠肝脏中DNA 6mA水平高于正常饮食(NCD)的小鼠(图1A)。
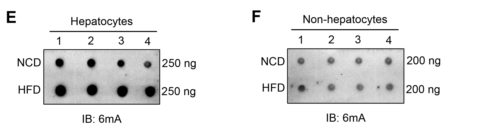
接着,作者从NCD和HFD喂养的小鼠中分离出肝细胞和非肝细胞。结果显示,与NCD小鼠相比,HFD小鼠肝细胞中DNA 6mA水平升高(图1E),而非肝细胞在NCD喂养的小鼠和HFD喂养的小鼠之间未观察到明显变化(图1F)。以上数据表明,在饮食诱导的脂肪肝肝细胞中DNA 6mA修饰水平增加。
2、DNA 6mA去甲基化酶ALKBH1在小鼠和人脂肪肝中的表达降低
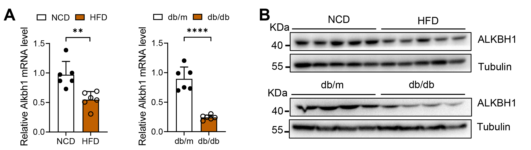
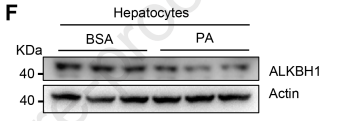
DNA 6mA修饰水平发生变化离不开修饰酶的作用,作者发现与NCD喂养的小鼠相比,HFD喂养的肥胖小鼠肝脏中ALKBH1的mRNA和蛋白水平显著下调(图2A-B)。另外,12周龄db/db遗传肥胖小鼠肝脏中ALKBH1的表达也降低(图2A-B)。
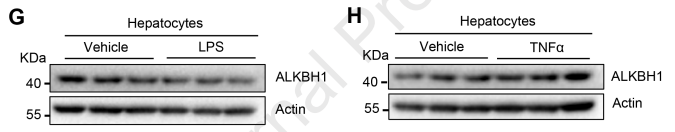
棕榈酸(PA)是一种常见的化学物质,用于刺激游离脂肪酸过量流入肝细胞,诱导脂质积累。作者发现,在PA处理后,ALKBH1的表达水平降低,但原代肝细胞中的脂多糖(LPS)和促炎细胞因子TNFα的表达水平并没有降低(图2F-H)。
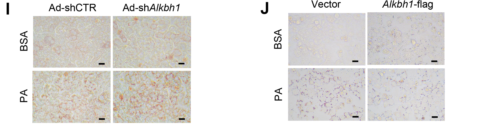
随后作者敲低ALKBH1可以促进PA诱导的脂滴积累,而过表达的ALKBH1则缓解了PA诱导的脂滴积累(图2I-J),表明ALKBH1在肝细胞脂质代谢中起着重要作用。
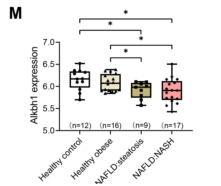
作者通过对公共数据库中数据集(GSE48452)进行分析,发现在肝脏脂肪变性和脂肪肝炎患者的肝脏样本中,ALKBH1的表达水平都有所下降,而在没有脂肪肝的健康肥胖受试者的肝脏中,ALKBH1的表达水平没有明显变化(图1M)。这些结果共同表明脂肪肝细胞中DNA 6mA去甲基化酶ALKBH1表达水平下降。
3、肝脏ALKBH1缺乏加重饮食诱导的肝脏脂肪变性和胰岛素抵抗
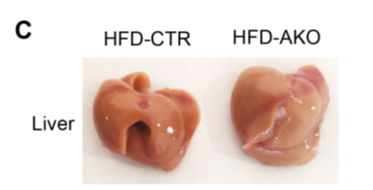
为了确定ALKBH1催化的DNA 6mA修饰是否在肝脏脂质代谢中发挥重要作用,作者建立肝脏特异性的ALKBH1敲除小鼠模型(AKO),如图3A。与对照组小鼠(未敲除ALKBH1)相比,NCD和HFD喂养的AKO小鼠的体重、食物摄入量和肝脂积累均正常,而其肝细胞内DNA 6mA修饰水平增加(图3B),AKO小鼠肝脏外观苍白,肝脏重量增加,HFD诱导的肝脏脂肪变性加剧(图4C)。

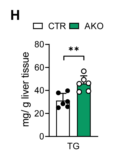
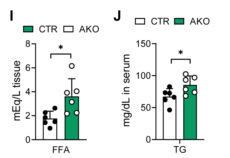
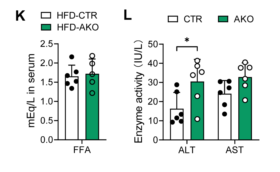
此外,HFD喂养的AKO小鼠肝脏TG(甘油三酸酯)、肝脏FFA(游离脂肪酸)和血清TG浓度也较高,但血清FFA不变(图4H-K),说明敲除ALKBH1加重了HFD诱导的肝损伤,表现为AKO小鼠血清ALT(丙氨酸氨基转移酶)和AST(天冬氨酸氨基转移酶)有升高的趋势(图4L)。
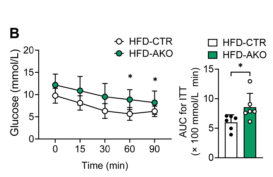
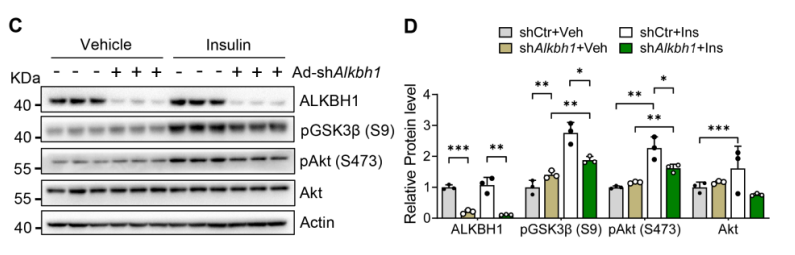
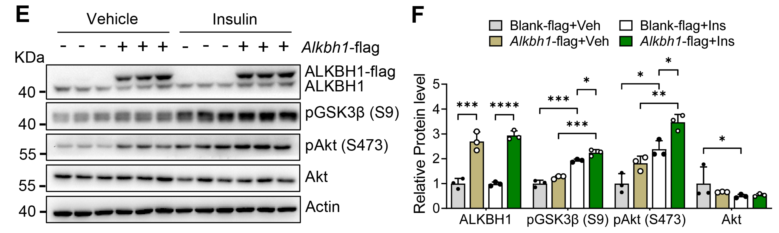
肝脏脂肪变性可导致糖耐量和肝脏胰岛素敏感性受损,敲除ALKBH1可以显著降低全身糖耐量(图5A)。与对照组小鼠相比,AKO小鼠的胰岛素抵抗也出现恶化(图5B),胰岛素诱导的Akt和GSK3β磷酸化在缺乏ALKBH1的原代肝细胞中得到缓解(图5C-D)。综上所述,肝脏特异性的ALKBH1对抑制饮食诱导的肝脏脂肪变性和胰岛素抵抗至关重要。

4、RNA-seq和ChIP-seq分析表明,ALKBH1调节脂质代谢
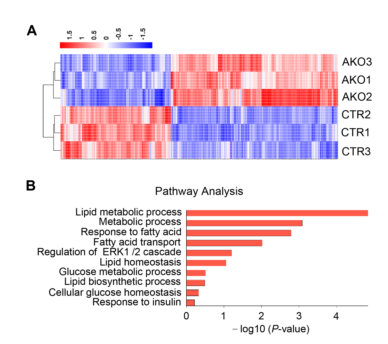
为了研究代谢应激下ALKBH1如何调节肝细胞中的脂肪沉积过程,作者对原代小鼠肝细胞进行了RNA-seq(图7A)。RNA-seq分析发现,AKO组中脂质代谢相关通路显著富集(图7B),表明ALKBH1在调节脂质代谢基因表达中发挥作用。
此外,与原代肝细胞的细胞质相比,ALKBH1蛋白在细胞核中的积累更多(图8A),表明ALKBH1可能主要在细胞核中作为DNA 6mA去甲基化酶发挥作用。为了进一步阐明ALKBH1调节肝细胞脂质代谢的机制,作者在过表达ALKBH1-flag融合蛋白的原代肝细胞中,通过康测科技金牌ChIP-seq,鉴定ALKBH1在肝细胞基因组中全局DNA的结合位点(图8B)。
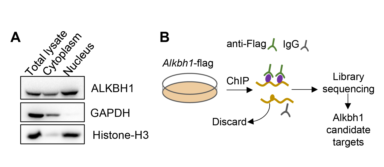
总的来说,在ALKBH1 IP样本中发现了8461个基因和8404个蛋白质编码基因。整合两组测序数据分析,得到437个ALKBH1潜在靶基因(图8D)。对上调的靶基因进行GO分析,也发现其在生物过程中显著富集,包括脂质代谢过程、脂质响应、脂肪酸运输和脂质生物合成(图8E)。
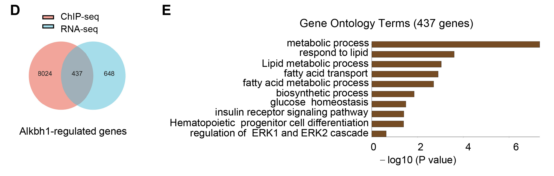
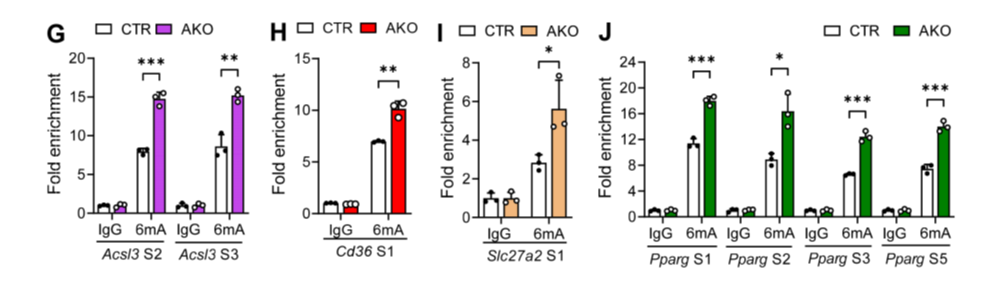
值得注意的是,在肝细胞中敲除ALKBH1后,脂质摄取基因Cd36、Slc27a2和脂质合成基因Ppar、Acsl3的表达水平显著增加,这些属于ALKBH1结合的靶标基因(通过ChIP-seq与RNA-seq取交集),而对其他脂质代谢相关基因的表达影响不大(图9A)。
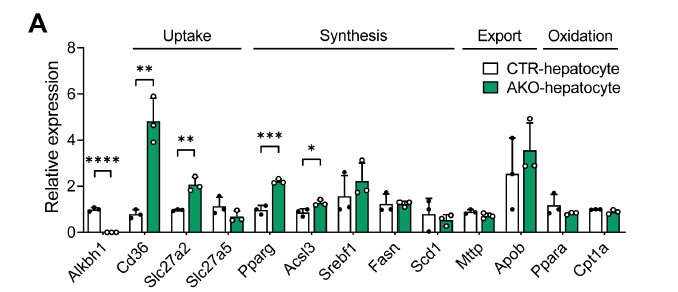
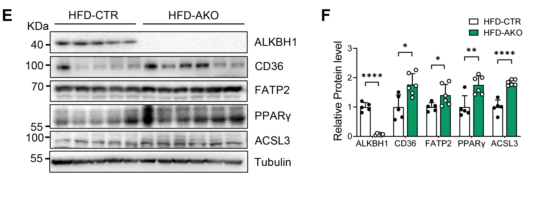
当NCD喂养时,AKO小鼠与对照组小鼠相比,这些脂质摄取和合成基因的转录略有增加。当进行HFD喂养时,这种影响更加明显,表现为AKO小鼠肝脏Cd36、Slc27a2、Pparg和Acsl3基因的mRNA和蛋白质水平显著上调(图9E-F)。综上所述,ALKBH1缺乏通过增加肝脏脂质摄取和合成的基因表达,促进HFD诱导的肝脏脂肪变性。
5、ALKBH1直接结合并降低参与脂质摄取和合成的基因的DNA 6mA甲基化修饰水平
通过ChIP-seq检测ALKBH1结合区域,确定Slc27a2、Acsl3、Cd36和Pparg基因结构中所有被ALKBH1蛋白占据的结合位点(图10A-B)。
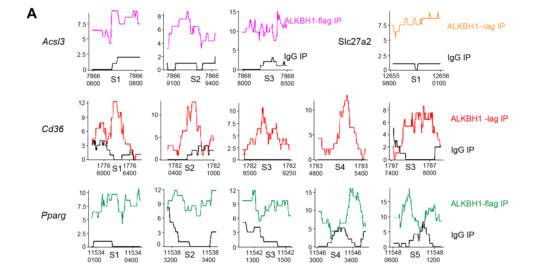
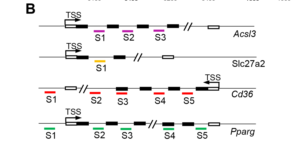
为了研究ALKBH1是否通过作为DNA 6mA去甲基化酶的功能来调节这些脂质代谢基因的表达,使用6mA抗体在野生型和ALKBH1缺失的原代肝细胞中进行6mA meDIP-qPCR。与对照肝细胞相比,AKO肝细胞中这些基因特异性结合位点上的6mA富集增强(图10G-J),导致基因表达增加。综上所述,ALKBH1通过在肝细胞的基因间区和/或内含子区去除这些基因的6mA修饰来调控Slc27a2、Acsl3、Cd36和Pparg的转录。
6、肝脏中过表达的ALKBH1对HFD诱导的肝脏脂肪变性和胰岛素抵抗具有保护作用
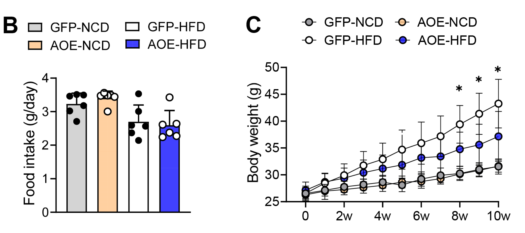
接下来,作者对小鼠肝脏通过腺相关病毒介导来过表达ALKBH1(AOE小鼠)进行功能增益分析。肝脏中过表达的ALKBH1显著缓解了HFD引起的体重增加、肝脏脂肪变性和肝脏重量,而不影响食物的摄入(图11B-E)。
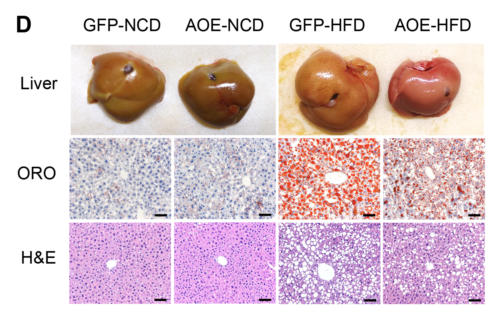
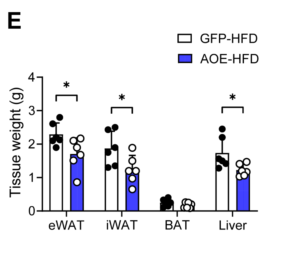
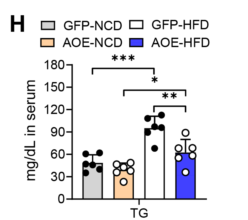
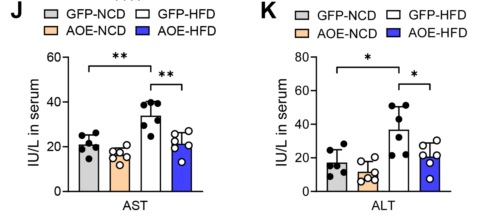
由此可见,过表达ALKBH1降低了HFD饲养条件下肝脏中TG和FFA的含量(图11F-G),同时也降低了血清中TG和FFA的循环水平(图11H-I),HFD喂养的AOE小鼠血清中AST和ALT的水平也低于对照组小鼠(图11J-K)。

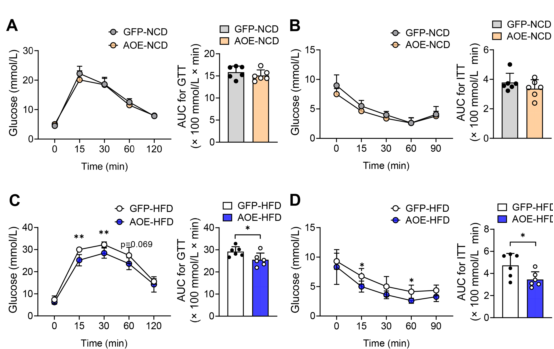
此外,肝脏中增强的ALKBH1表达不影响NCD小鼠的全身糖耐量和胰岛素敏感性(图12A-B),而显著改善HFD诱导的糖耐量和胰岛素抵抗(图12C-D)。
与对照组相比,在过表达ALKBH1的原代肝细胞中也观察到胰岛素诱导的Akt和GSK3β磷酸化的增加,从而改善了胰岛素敏感性(图12E-F)。
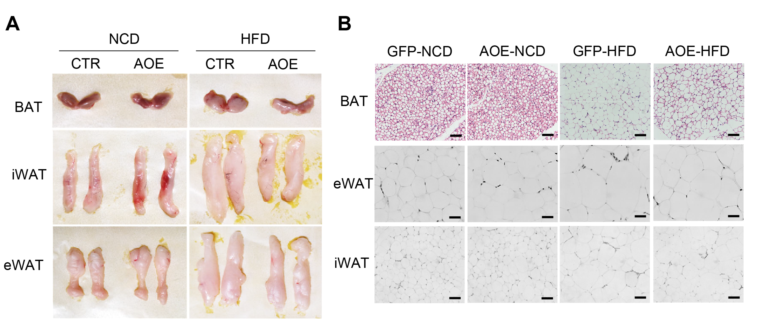
此外,在HFD条件下,与对照组小鼠相比,AOE小鼠的体重下降,脂肪组织体积和脂肪细胞大小减少(图13A-B)。
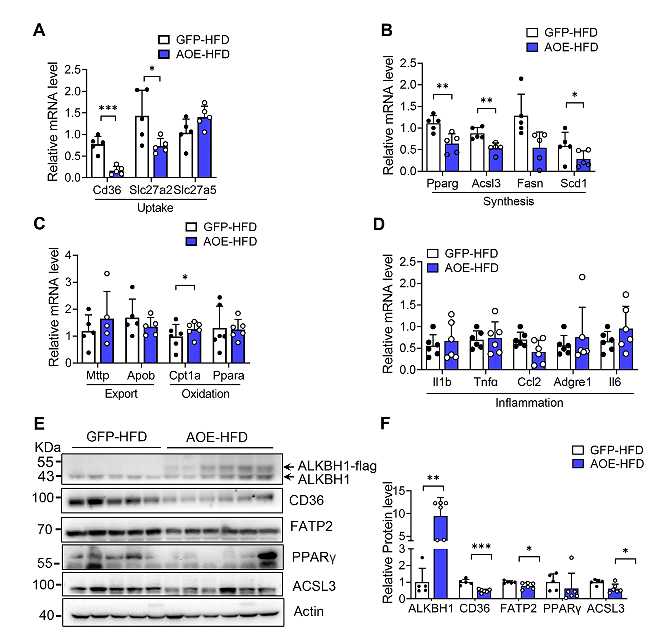
与通过敲除ALKBH1诱导的脂肪酸摄取和脂肪生成相反,在HFD喂养的AOE小鼠的肝脏中,Cd36、Slc27a2、Pparg和Acsl3基因的表达在mRNA和蛋白质水平上都受到抑制(图14A-F)。综上所述,肝脏ALKBH1表达水平升高可以通过减少脂质摄入和生物合成来保护HFD诱导的肝脏脂肪变性和胰岛素抵抗。
#
研究总结
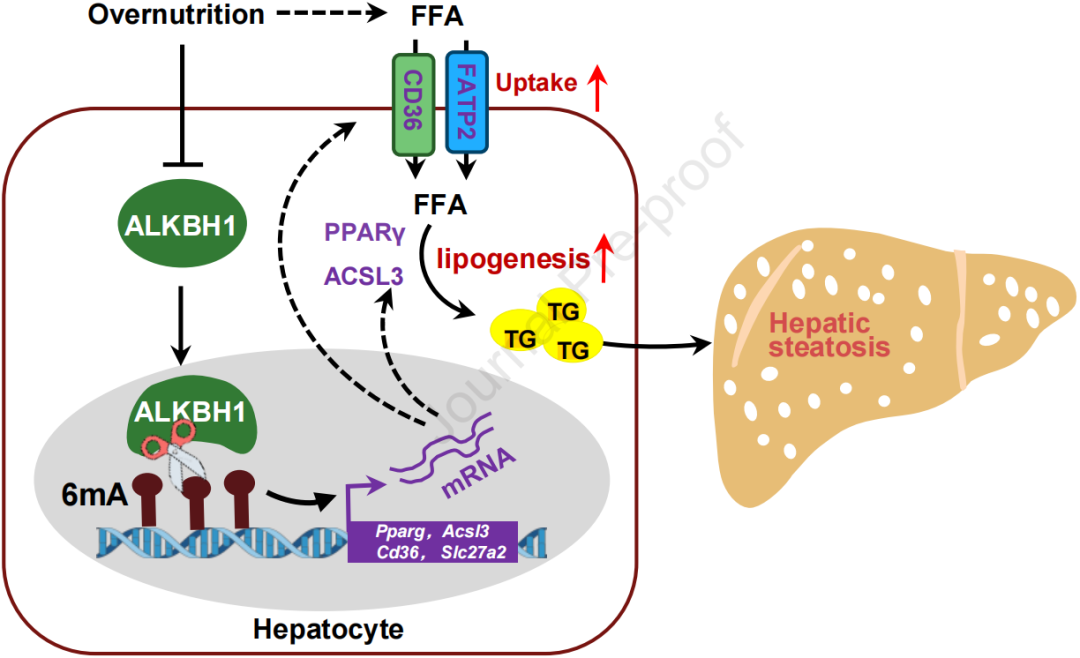
1、饮食诱导的高脂小鼠肝脏中DNA 6mA水平高于正常小鼠;
2、HFD喂养的肥胖小鼠和遗传性肥胖小鼠的肝脏中ALKBH1的mRNA和蛋白水平显著下调。敲低肝细胞ALKBH1可以促进棕榈酸诱导的脂肪滴积累,而过表达的ALKBH1则可以缓解这一过程;
3、正常饮食条件下,敲除ALKBH1(AKO)的小鼠的肝细胞内DNA 6mA水平增强;
4、RNA-seq和ChIP-seq分析表明,敲除肝细胞中ALKBH1后,与脂质摄取基因和脂质合成相关的ALKBH1靶标基因的表达显著增加,可以促进HFD诱导的肝脏脂肪变性;
5、过表达ALKBH1可以抑制脂质摄取和合成,缓解饮食诱导的肝脏脂肪变性和胰岛素抵抗,说明ALKBH1作为DNA 6mA修饰的表观遗传抑制因子在肝脏脂肪酸代谢中不可或缺的作用,并为脂肪肝提供了潜在的治疗靶点。